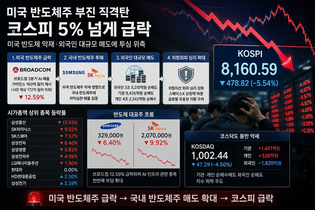













영아와 아이들에서 중증 근력 약화를 유발하는 유전성 질환인 제 1형 척수성 근위축증(SMA-1: Spinal Muscular Atrophy-1)은 현재 까지 치료제가 없다.
그러나 9일 올랜도 Nemours 소아병원 연구팀이 '란셋'지에 밝힌 임상시험 연구결과에 의하면 뉴시너센(nusinerse n) 이라는 새로운 약물이 SMA-1 진단을 받은 영아에서 효과적인 것으로 나타났다.
SMA-1 은 미국에서만 매 년 250명의 신생하가 앓는 질환으로 Werdnig-Hoffmann 질환으로도 불리며 영아들이 양 부모로부터 SMN1 이라는 변이 유전자를 유전받을 시 발병한다.
SMA-1 는 영아들에서 가장 흔한 사망의 유전적 원인으로 예후는 매우 좋지 않아 진단 2년내 대부분의 영아들이 사망한다.
SMN1 유전자는 정상적으로는 survival motor neuron (SMN) 이라는 단일 단백질을 코딩하는 바 이 같은 단백질은 척추로 부터 근육으로 가는 신경을 보호하며 이 같은 단백질이 없을 경우 신경은 퇴화해 비가역적인 근위축이 유발된다.
하지만 이 같은 결함 유전자를 가진 사람들은 백업 유전자나 SMN1 유전자의 정상 카피본을 가지고 있어 증상 없이 살 수 있지만 SMA-1 을 앓는 영아들은 하나가 아닌 두 개의 제대로 기능하지 않는 SMN1 을 유전받아 근위축이 유발 결국 삼킬수도 없고 심지어 호흡할수도 있다.
연구팀에 의하면 SMN1 유전자가 운동 신경 단백질을 생성할 수 있는 유전자 코드의 한 섹션이 있는 바 뉴시너넨이라는 새로운 약물이 결함이 있는 RNA sequence 에 결합 기능을 하는 SMN 단백질을 생성하게 하는 방식으로 해당 유전자를 재구성할 수 있는 것으로 나타났다.
연구팀은 "임상 시험 결과 뉴시너렌이 효과적이고 안전하며 환자의 순응도가 높은 것으로 나타났으며 FDA가 현재 승인 검토 중에 있다"라고 밝혔다.
그러나 9일 올랜도 Nemours 소아병원 연구팀이 '란셋'지에 밝힌 임상시험 연구결과에 의하면 뉴시너센(nusinerse n) 이라는 새로운 약물이 SMA-1 진단을 받은 영아에서 효과적인 것으로 나타났다.
SMA-1 은 미국에서만 매 년 250명의 신생하가 앓는 질환으로 Werdnig-Hoffmann 질환으로도 불리며 영아들이 양 부모로부터 SMN1 이라는 변이 유전자를 유전받을 시 발병한다.
SMA-1 는 영아들에서 가장 흔한 사망의 유전적 원인으로 예후는 매우 좋지 않아 진단 2년내 대부분의 영아들이 사망한다.
SMN1 유전자는 정상적으로는 survival motor neuron (SMN) 이라는 단일 단백질을 코딩하는 바 이 같은 단백질은 척추로 부터 근육으로 가는 신경을 보호하며 이 같은 단백질이 없을 경우 신경은 퇴화해 비가역적인 근위축이 유발된다.
하지만 이 같은 결함 유전자를 가진 사람들은 백업 유전자나 SMN1 유전자의 정상 카피본을 가지고 있어 증상 없이 살 수 있지만 SMA-1 을 앓는 영아들은 하나가 아닌 두 개의 제대로 기능하지 않는 SMN1 을 유전받아 근위축이 유발 결국 삼킬수도 없고 심지어 호흡할수도 있다.
연구팀에 의하면 SMN1 유전자가 운동 신경 단백질을 생성할 수 있는 유전자 코드의 한 섹션이 있는 바 뉴시너넨이라는 새로운 약물이 결함이 있는 RNA sequence 에 결합 기능을 하는 SMN 단백질을 생성하게 하는 방식으로 해당 유전자를 재구성할 수 있는 것으로 나타났다.
연구팀은 "임상 시험 결과 뉴시너렌이 효과적이고 안전하며 환자의 순응도가 높은 것으로 나타났으며 FDA가 현재 승인 검토 중에 있다"라고 밝혔다.
메디컬투데이 강현성 ([email protected])

[저작권자ⓒ 메디컬투데이. 무단전재-재배포 금지]